
Issue #1: April, 2026

Editor’s Note
Welcome to Issue 1. The intent of this newsletter is simple: a monthly, opinionated pass through the PDAC literature and news cycle, with enough editorial framing to help you decide what is worth reading in full and what you can safely skim. I will not attempt comprehensive coverage but will try to identify what matters, say why, and flag the caveats worth considering.
April 2026 is an unusually substantive month to debut. Within a single 24-hour window on April 13-14, two readouts in metastatic PDAC landed with hazard ratios that most of us have not seen in this disease in our careers: daraxonrasib (RMC-6236) in the phase 3 RASolute 302 trial, and elraglusib in the phase 2 Actuate 1801 trial, published in Nature Medicine. Both roughly doubled one-year survival. A third paper that deserves the same attention, the POLAR trial of pembrolizumab plus olaparib maintenance in HRD biomarker-selected metastatic PDAC, was published online in Nature Medicine in late March and is landing its full impact in the April cycle. None of these will cure anyone, and all three carry real caveats worth considering. But three distinct positive signals in metastatic PDAC in a single month is itself the story.
AACR 2026 in San Diego ran April 17-22, overlapping publication of this issue. I will cover the meeting properly in its own entry. Below, I flag a few sessions and company presentations worth watching as the abstracts become public.
— Ryan
Clinical Trials & Practice
Daraxonrasib (RMC-6236) phase 3 RASolute 302: targeted therapy finally works in metastatic PDAC
On April 13, Revolution Medicines released topline results from RASolute 302 (NCT06625320), a global randomized phase 3 trial of daraxonrasib, an oral RAS(ON) multi-selective inhibitor, versus investigator’s-choice cytotoxic chemotherapy in previously treated metastatic PDAC. In the intent-to-treat population, median overall survival was 13.2 months with daraxonrasib compared with 6.7 months with chemotherapy, with a hazard ratio of 0.40 (p < 0.0001). Both co-primary endpoints, PFS and OS in the RAS G12-mutant population, were also met. Safety was consistent with prior reports (predominantly low-grade rash and GI toxicity). Revolution intends to submit for approval via the FDA’s Commissioner’s National Priority Voucher pathway, and detailed data are scheduled for ASCO 2026.
Why this matters. An HR of 0.40 in second-line metastatic PDAC is not a number we are used to seeing. For context, the chemotherapy comparator arm performed exactly as historical second-line data would predict (median 6.7 months), so the daraxonrasib effect is not an artifact of an underperforming control. The mechanism is also noteworthy: unlike allele-specific KRAS G12C agents (sotorasib, adagrasib), daraxonrasib is a tri-complex inhibitor that binds cyclophilin A and then engages the active GTP-bound state of both mutant and wild-type RAS, which is what allows it to act across G12D, G12V, G12R, and wild-type tumors. For a disease where 90%+ of tumors are RAS-driven and the variant distribution is heterogeneous, broad-spectrum RAS targeting is conceptually the right drug for the biology.
Worth considering.
These are press release numbers. The full data, including the G12-mutant primary analysis, subgroup performance, duration of response, and quality-of-life outcomes, are not yet public.
Second-line is a specific context. The front-line question, where chemotherapy still has meaningful activity and the competitive landscape is more crowded, is what will define daraxonrasib’s eventual role. The relevant combination trials are ongoing.
The ITT population includes both RAS-mutant and RAS wild-type patients, and the company has emphasized broad activity across subtypes. Parse the subgroup results carefully when they are presented at ASCO.
Bottom line. Assume this drug is coming, and start thinking now about how it integrates into our sequencing, molecular testing workflows, and adjuvant trial landscape.
Revolution Medicines press release (April 13, 2026): ir.revmed.com
OncLive summary: onclive.com
Elraglusib plus GnP in first-line metastatic PDAC (phase 2, Nature Medicine)
Published in Nature Medicine on April 14, the open-label phase 2 Actuate 1801 trial (NCT03678883) randomized 233 treatment-naive patients with metastatic PDAC 2:1 to weekly elraglusib with gemcitabine and nab-paclitaxel (GnP) versus GnP alone. Median overall survival was 10.1 months in the combination arm versus 7.2 months with chemotherapy alone (HR 0.63). One-year survival rate was 44% versus 22%. Grade 3+ toxicity was dominated by neutropenia (52% vs 31%) and fatigue (17% vs 5%). Exploratory correlatives showed higher baseline CXCL2 and TRAIL ligand levels associated with benefit in the elraglusib arm, and treatment was associated with expansion of intratumoral cytotoxic immune populations. A phase 3 trial is planned.
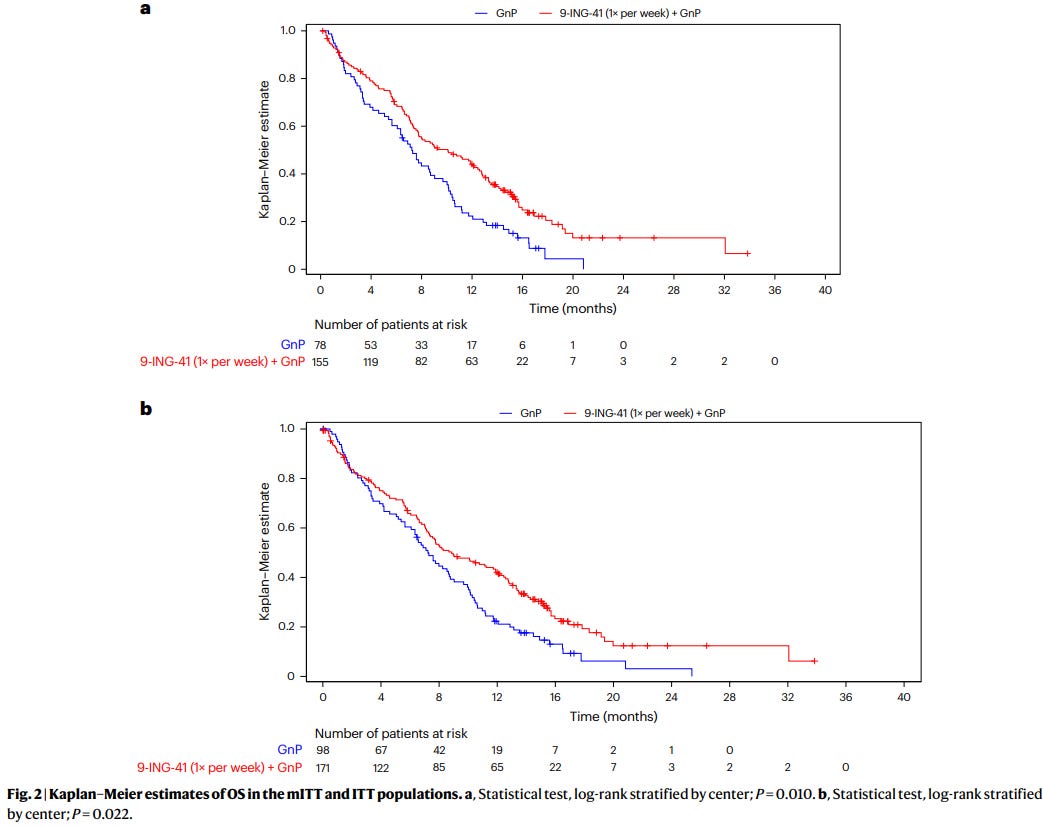
Why this matters. First-line metastatic PDAC has seen almost no positive randomized readouts in the last decade (GnP + name your drug). A three-month median OS improvement with a manageable toxicity profile, if it holds in phase 3, would represent a meaningful shift in the front-line standard. The immunologic correlatives are also interesting and align with the narrative that elraglusib’s effect is microenvironment-mediated rather than purely cytotoxic.
Worth considering.
This is phase 2, not phase 3. The 2:1 randomization, modest sample size, and open-label design all create room for the effect to shrink on replication.
The mechanism is not settled. Elraglusib was developed as a GSK-3β inhibitor, and the Nature Medicine paper frames the biology that way. However, a bioRxiv preprint (Walz et al., 2024) provided pharmacologic and genetic evidence that elraglusib’s cytotoxic activity is driven by microtubule destabilization, not GSK-3β inhibition, with chemical-genetic dissociation from GSK3A/B knockdown. If true, this has implications for rational combination design and biomarker development. Whatever the true target, the clinical signal in 1801 is the signal. But when the Nature Medicine paper and a bioRxiv preprint disagree about mechanism, we should be honest that the on-target biology is unresolved. Perhaps our very own in-house expert, Dan Billadeau, could share his opinion.
Benefit over current front-line mFOLFIRINOX, which most of us use for fit patients, is not addressed by this trial. The comparator is GnP alone.
Bottom line. Promising, and the immune correlatives are provocative. Wait for the phase 3. Keep an eye on mechanism-of-action work.
Chidharla et al, Nat Med (April 14, 2026): nature.com/articles/s41591-026-04327-4
Walz et al, bioRxiv (mechanism critique): biorxiv.org/content/10.1101/2024.07.06.602326
Taken together: the shape of targeted therapy in PDAC
It is worth naming what these two readouts mean together. In a single week, we got: (1) a broad-spectrum RAS inhibitor doubling OS in second-line, and (2) a microenvironment-modulating agent (whatever its actual target) doubling one-year survival in first-line. Neither is curative. Both are consistent with a view of PDAC where meaningful gains come from simultaneously hitting dominant oncogenic drivers and disarming the supportive stromal-immune ecology. That is not a new conceptual framework. It is the first time in a while that the clinical readouts have been consistent with it.
POLAR: pembrolizumab plus olaparib maintenance in HRD biomarker-selected metastatic PDAC (Park et al., Nature Medicine)
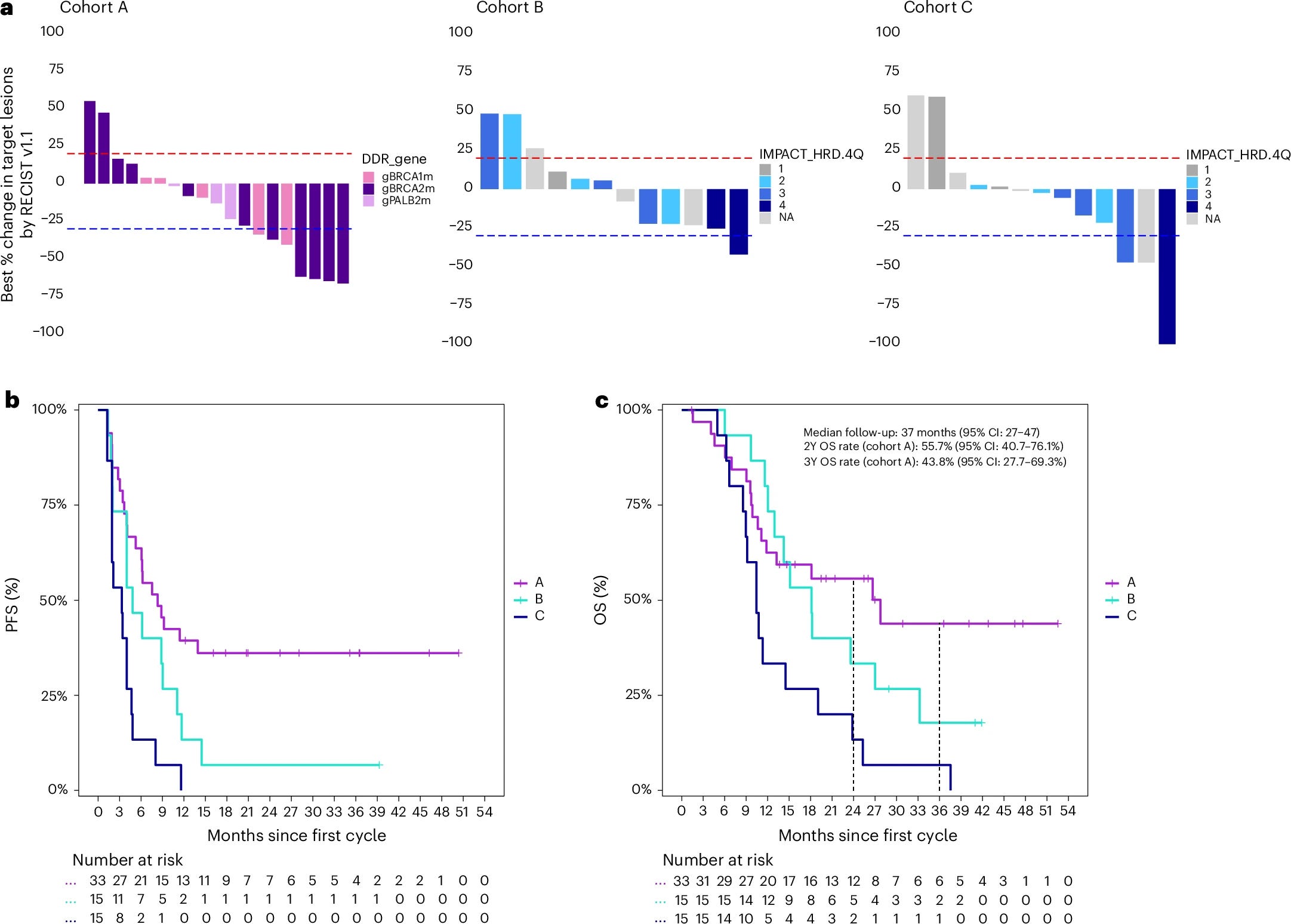
Published online March 25, but a defining paper for this cycle. POLAR (NCT04666740) is a phase 2, single-center (MSKCC), non-randomized basket trial of pembrolizumab plus olaparib maintenance following response to platinum-based induction in 63 patients with metastatic PDAC, stratified into three biomarker-defined cohorts: cohort A (n = 33), core HRD defined as pathogenic germline or somatic BRCA1, BRCA2, or PALB2 mutations; cohort B (n = 15), non-core HRD mutations (ATM, CHEK2, MUTYH, BLM, FANCC, and others); and cohort C (n = 15), HRD wild-type with at least 6 months of platinum sensitivity. All participants received olaparib 300 mg BID plus pembrolizumab IV every 3 weeks for 6 months and then every 6 weeks until progression or unacceptable toxicity.
The trial did not meet its prespecified co-primary endpoints in cohort A, which required an ORR of at least 43% and a 6-month PFS rate of at least 77%. Observed values were ORR 35% (7/20 RECIST-evaluable; 95% CI 15-59%) and 6-month PFS rate 64% (95% CI 49-82%). The authors acknowledge that the bar was intentionally aggressive for a non-registrational single-center study.
That said, the downstream clinical signal is meaningful. In cohort A at median follow-up of 37 months, median PFS was 8.3 months, median OS was 28 months, 2-year OS was 56% (95% CI 41-76%), and 3-year OS was 44% (95% CI 28-69%). For historical context, the olaparib monotherapy arm in POLO (the registrational germline BRCA-mutated PDAC maintenance trial) reported ORR 23%, 6-month PFS 53%, 2-year OS 37%, and 3-year OS 34%. POLAR cohort A numerically exceeds POLO on every clinical metric. Cohort B (ncHRD) and cohort C (HRP, platinum-sensitive) did progressively worse: median OS 18 and 10 months, respectively, supporting the biomarker stratification logic. A subgroup analysis within cohort A showed BRCA2 and PALB2 patients did numerically better than BRCA1 patients on both PFS and OS, though sample sizes are small.
The translational work is what elevates this paper beyond the clinical numbers. HRD tumors (cohort A) had significantly higher WES-derived TMB, IMPACT-HRD scores, indel burden, and frameshift indel burden compared to HRP tumors (cohort C), consistent with genomic instability producing an enriched neoantigen substrate, particularly the high-quality frameshift-derived neoantigens. H&E-derived TIL density and CD3+CD8+ T cell infiltration by mIF were also higher in cohort A. Critically, neoantigen burden correlated with IMPACT-HRD score (R = 0.48, p = 0.008) but not with CD8+ T cell infiltration (p = 0.95). The authors interpret this, correctly I think, as antigen recognition being necessary but not sufficient for productive immune engagement in PDAC, with residual barriers such as CAF-mediated T cell exclusion and KRAS G12D-driven myeloid suppression persisting even in neoantigen-rich HRD tumors. ctDNA mRD negativity at baseline and 6 weeks was associated with durable benefit, which is biologically coherent and practically actionable for trial design.
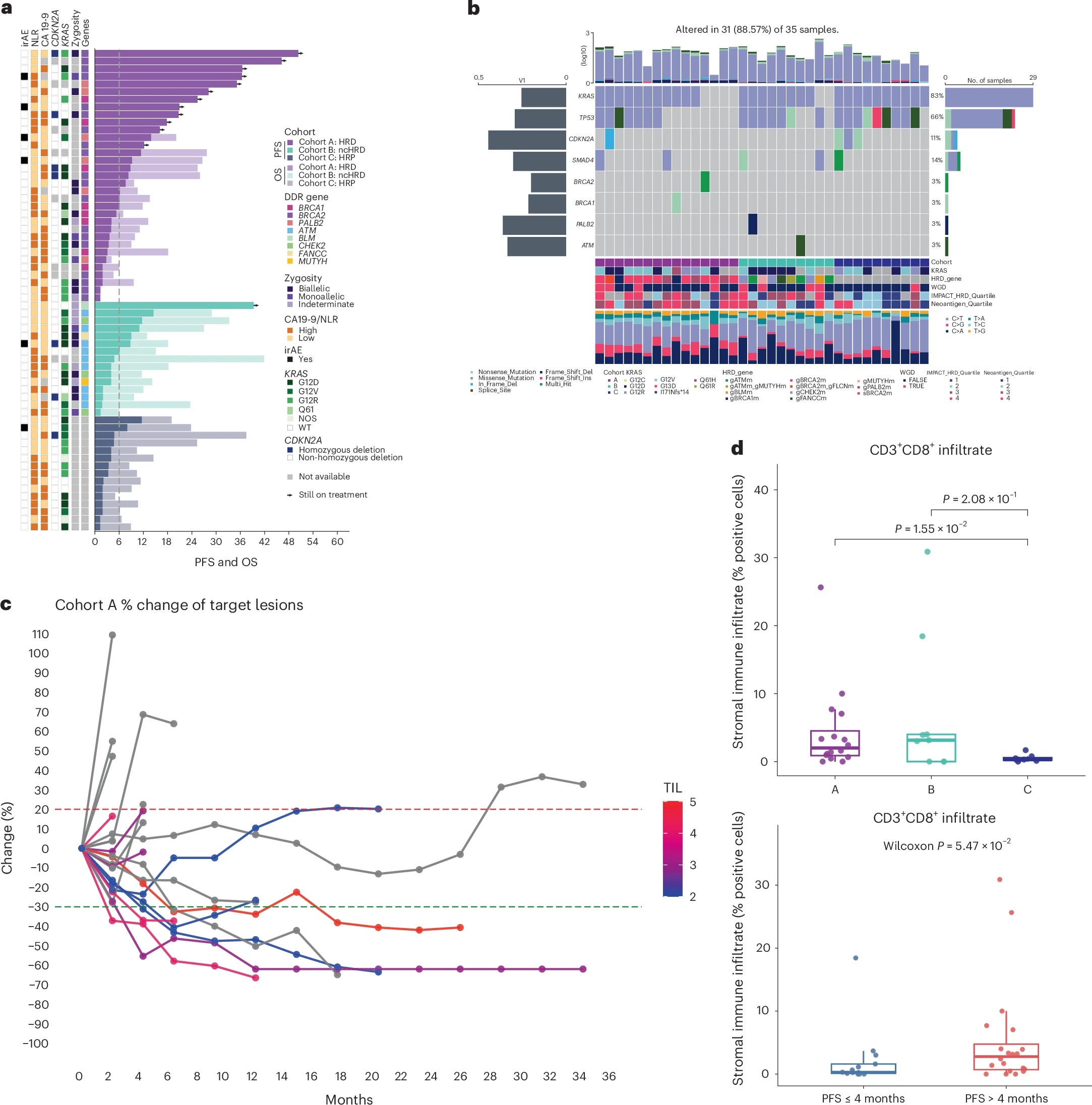
Safety was as expected for the combination, with no grade 4 or 5 TRAEs. Grade 3 anemia occurred in 15% overall, and there were a small number of grade 3 irAEs (pneumonitis, colitis).
Why this matters. This is the first prospective dataset supporting combined PARPi-immunotherapy maintenance in biomarker-selected HRD metastatic PDAC, and the durable survival signal in cohort A is real. Equally important, POLAR advances the conceptual story in a specific way: HRD biology provides an immunogenic substrate that is necessary but not sufficient, and the next round of trials needs to pair DDR-based biomarker selection with interventions that address the residual exclusionary TME features.
Worth considering.
Single-center, single-arm, non-randomized. The favorable comparison to POLO is historical, not prospective.
The prespecified co-primary endpoints were not met. Framing the trial as positive requires leaning on secondary endpoints, which is scientifically legitimate here but should be named clearly in any discussion.
Only 45% of cohort A (15/33) had evaluable tissue zygosity for biallelic loss determination. The authors attribute this to low tumor cellularity from profound platinum response, which is itself an enrollment-driven source of selection bias.
Cohort B (ncHRD) and cohort C outcomes were modest, tempering enthusiasm for broader biomarker expansion beyond core HRD without additional selection.
Randomized confirmation is needed. SWOG S2001 (NCT04548752) is the relevant ongoing trial.
On the choice of agents. POLAR also deserves reading against Reiss et al. (Lancet Oncol 2022), the randomized phase 1b/2 trial from Penn that compared niraparib plus nivolumab versus niraparib plus ipilimumab as maintenance in 91 patients with platinum-sensitive advanced PDAC (selected on platinum sensitivity, not HRD biomarkers). The niraparib plus ipilimumab arm met its primary endpoint with 6-month PFS 59.6% and median PFS 8.1 months. The niraparib plus nivolumab arm was inferior, with 6-month PFS 20.6% and median PFS 1.9 months. Two implications follow for POLAR. First, CTLA-4 blockade appears to outperform PD-1 blockade when paired with a PARPi in PDAC, which is biologically coherent: the rate-limiting step in PDAC’s immune response is priming and infiltration, not effector exhaustion, and ipilimumab acts on the former (repertoire expansion, Treg modulation) while pembrolizumab and nivolumab act on the latter. POLAR’s own finding that neoantigen burden does not correlate with CD8+ infiltration is consistent with this, and it implies that the specific choice of pembrolizumab may not be optimal. Second, niraparib is more PARP1-selective than olaparib, and next-generation PARP1-selective agents, which POLAR’s authors name as a near-term direction in their own discussion, may produce more neoantigen-generating DNA damage and more robust cGAS-STING activation. A rational successor to POLAR in HRD PDAC would likely pair a CTLA-4-directed agent, or a combined CTLA-4 plus PD-1 regimen, with a more potent or PARP1-selective PARPi. POLAR validates the PARPi-immunotherapy maintenance framework in HRD PDAC. The specific agent choices may not be the optimal ones.
Bottom line. Not practice-changing on its own because the randomized data are not here yet, but directionally supportive of PARPi-immunotherapy as a maintenance strategy in core HRD PDAC, and a rigorous translational framework for future biomarker-integrated immunotherapy trial design in this disease. SWOG S2001 will test whether the POLAR signal replicates. The longer-term question, informed by Reiss, is whether pembrolizumab plus olaparib is even the right pairing to be testing.
Park et al, Nat Med (25 March 2026): DOI: 10.1038/s41591-026-04299-5
Reiss et al, Lancet Oncol 2022;23:1009-1020: DOI: 10.1016/S1470-2045(22)00369-2
Translational Science
A four-marker plasma panel for early PDAC detection (Krusen et al., Clin Cancer Res)
A Penn–Mayo collaboration led by Kenneth Zaret (Penn) and including Shounak Majumder, Ann Oberg, and William Bamlet from Mayo Rochester published validation of an expanded plasma biomarker panel for early PDAC detection. The panel combines CA19-9, THBS2, aminopeptidase N (ANPEP), and polymeric immunoglobulin receptor (PIGR). Across two retrospective phase II cohorts (537 Mayo, 135 Penn plasma samples, 672 total), the four-marker panel achieved AUC of 0.87 for stage I/II versus disease controls and 0.91 for stage I-IV versus disease controls in the Mayo cohort. At 95% specificity, the panel detected 91.9% of all-stage PDAC and 87.5% of stage I/II disease.
Why this matters. CA19-9 alone is limited by Lewis antigen nonexpression (roughly 10-15% of patients) and elevation in benign conditions. Adding THBS2 helped modestly. ANPEP and PIGR are mechanistically distinct (aminopeptidase activity and transcytosis of polymeric immunoglobulins, respectively), and the AUC gains in stage I/II detection are what actually matters for this disease. This is now a credible candidate for prospective evaluation in high-risk surveillance cohorts.
Worth considering.
These are retrospective phase II validation cohorts. The pre-diagnostic performance, which is what determines utility as a screening tool, is not yet established and will require prospective cohort evaluation.
The Mayo cohort is substantially larger than the Penn cohort, and both are enriched for confirmed cases versus a true screening population. Performance in a true high-risk surveillance setting (e.g., CAPS-eligible individuals) will be lower.
This test is not yet clinically available. Do not try to order ANPEP through Epic today.
Bottom line. The most promising plasma-based early detection effort I have seen this year, with Mayo leadership. Watch for prospective validation designs.
Krusen et al, Clin Cancer Res 2026;32(4):756: DOI: 10.1158/1078-0432.CCR-25-3297
NIH news summary: nih.gov/news-events
Basic Science
CAF subtype-specific CRISPR perturbations in PDAC coculture (Saputra et al., Mol Oncol)
Worth a read if CAF biology is adjacent to your work. The group developed immortalized CAF cell lines from patient-derived PDAC and used single-cell transcriptomics plus CRISPR perturbations to assess subtype-specific effects of candidate stromal targets. Two points stand out: (1) the model captures interconvertibility between CAF states rather than treating them as stable categories, which aligns with mounting evidence that iCAF/myCAF/apCAF designations are poles on a continuum rather than discrete cell types, and (2) CRISPR perturbations had subtype-specific effects, meaning that a “CAF-targeting” therapeutic strategy without subtype resolution is likely to produce inconsistent phenotypes.
Why this matters. CAF biology has been a graveyard of clinical programs (recall the hedgehog inhibitor results, and the more recent FAP-targeted approaches). One reason is that aggregate CAF targeting is biologically incoherent when CAF populations contain both tumor-promoting and tumor-restraining subtypes. Work like this, which approaches CAFs with subtype-specific resolution, is how we get to selective therapeutic vulnerabilities.
Worth considering. In vitro coculture, immortalized lines. The next step is validation in orthotopic models and ideally in spatially resolved patient tissue. Translatability is acknowledged as an open question by the authors themselves.
Saputra et al, Mol Oncol (2026): DOI: 10.1002/1878-0261.70153
KRAS G12R as allele-specific biology (Iacobuzio-Donahue commentary in Cancer Research, on Burge et al.)
Christine Iacobuzio-Donahue’s mid-April commentary in Cancer Research, “Altered PI3K and ERK/MAPK Signaling in KRAS G12R-Driven Pancreatic Cancer Presents Opportunities for Precision Therapy,” accompanies new primary data from the Hobbs lab at MUSC (Burge et al., Cancer Research 2026). The underlying paper develops a KrasG12R/Tp53R172H GEMM and compares it head-to-head with matched KrasG12D models. Three findings are worth noting.
First, G12R GEMMs show strikingly limited tumorigenesis compared to G12D counterparts: only about 10% develop pancreatic tumors at one year versus rapid multifocal disease in G12D mice. G12R and G12D activate similar pancreas-specific transcriptional networks, but G12R promotes them less robustly, and ERK/MAPK nuclear translocation is reduced. This fits the well-established clinical observation that patients with G12R-mutant PDAC have better OS than patients with G12D/V mutations.
Second, G12R tumors have a distinctive TME with reduced collagen deposition and reduced metastatic liver invasion. This is a phenotype worth dwelling on: the driver allele appears to shape not only intrinsic cell behavior but the stromal architecture and metastatic niche colonization. Different KRAS alleles produce different tumor ecologies, not just different cell-autonomous signaling.
Third, the authors show that KRAS is not the primary driver of PI3K activity in human PDAC regardless of mutation allele. Rather, PTEN inactivation through a redox-mediated modification supports elevated basal PI3K signaling across PDAC. All four Class I PI3K isoforms contribute, and no single isoform-selective inhibitor is sufficient. This reframes the PI3K-targeting problem: even in G12R tumors where KRAS cannot directly activate p110α, the pathway is active through KRAS-independent mechanisms.
Why this matters. The commentary frames G12R as both prognostic (confirmed) and potentially a therapeutic stratifier. Prior work from the same group (Hobbs et al., Cancer Discov 2020) showed G12R-mutant PDAC preferentially sensitive to MEK/ERK inhibition and to combined ERK plus autophagy inhibition (chloroquine). The Burge GEMM data provide mechanistic grounding for why. The broader implication is that KRAS-mutant PDAC is not biologically homogeneous, and trial designs that pool alleles under a single “RAS-mutant” bucket may average over meaningful biology. This is directly relevant to how we should interpret the daraxonrasib readout: the ITT population includes G12D, G12V, G12R, and wild-type disease, and the agent’s performance in each allele class is worth watching when the detailed RASolute 302 data present at ASCO.
Worth considering. GEMM data, not clinical. The reduced tumorigenicity in G12R GEMMs does not translate to “G12R is indolent” in patients; these tumors still kill, and the survival advantage over G12D/V is modest in absolute terms. The PTEN-PI3K finding adds biological complexity to the targeting question rather than simplifying it.
Iacobuzio-Donahue, Cancer Res 2026;86(8):1817 (commentary): aacrjournals.org
Burge et al, Cancer Res 2026 (primary paper): DOI: 10.1158/0008-5472.CAN-25-2630
Foundational: Hobbs et al, Cancer Discov 2020;10:104: DOI: 10.1158/2159-8290.CD-19-1006
Meeting Highlights: AACR 2026 Preview
AACR Annual Meeting 2026 runs April 17-22 at the San Diego Convention Center. Because this issue lands while the meeting is still live, full coverage is not possible here (plus, this is getting lengthy). A dedicated “Special Report: AACR 2026” bulletin will go out within 7-10 days of the meeting’s close, covering the PDAC-relevant content with the same editorial lens as the monthly issues. That approach gives meeting coverage the attention it warrants without compromising the end-of-month cadence for regular issues.
For now, a few items on my watch list:
“Advances in Pancreatic Cancer Research and Treatment” plenary, chaired by Anirban Maitra (NYU Langone Perlmutter). Major session.
ClearNote Health is presenting updated Avantect Pancreatic Cancer Test data with reported sensitivity of 82.6% and specificity of 97.5% in the elevated-risk population, plus an epigenomic liquid biopsy study (Golan et al, Sheba) for platinum/PARP response prediction in germline BRCA-associated PDAC. Early detection and response monitoring both.
Stromal and 3D organoid biology. UF Health’s poster set includes a tunable 3D organoid model linking matrix stiffness to stromal activation and cytokine signaling (Ruiz-Rivera et al), and a coculture study on ECM stiffness and glutamine availability in PDAC-CAF metabolic crosstalk (Barajas et al). Relevant to the broader conversation about stromal targeting.
Immunotherapy in cold tumors. Leukogene Therapeutics is presenting data on MHC class II-engager bispecifics in PDAC and AML. The concept of leveraging MHC II rather than MHC I in immunologically cold tumors is worth tracking.
If you are at the meeting and see something that should make the Special Report, send me a note.
AACR Annual Meeting 2026 program: aacr.org
Housekeeping
This newsletter is intended for the Mayo Pancreatic Cancer Working Group. If you have items you think should be included in Issue 2, or disagree with anything above, reply directly. Corrections welcome and expected.
Ryan Carr, MD, PhD
Assistant Professor of Oncology
Mayo Clinic Rochester